
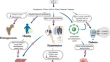

EPMA Journal publishes results and achievements in primary (individualised protection of healthy persons against health-to-disease transition) and secondary (individualised protection against disease progression) care

Predictive, Preventive and Personalised Approach in Primary and Secondary Healthcare; Special issue – Internationally awarded research innovation


View the latest Editor-in-Chief highlights from EPMA Journal.

View the latest Editor-in-Chief highlights from EPMA Journal.
View the latest Editor-in-Chief highlights from EPMA Journal.
© European Association for Predictive, Preventive and Personalised Medicine (EPMA)